

doi herehttps://doi.org/10.1021/acs.analchem.6b02082
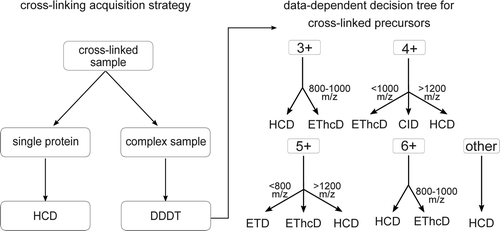
Abstract goes hereCross-linking/mass spectrometry has evolved into a robust technology that reveals structural insights into proteins and protein complexes. We leverage a new tribrid instrument with improved fragmentation capacities in a systematic comparison to identify which fragmentation method would be best for the identification of cross-linked peptides. Specifically, we explored three fragmentation methods and two combinations: collision-induced dissociation (CID), beam-type CID (HCD), electron-transfer dissociation (ETD), ETciD, and EThcD. Trypsin-digested, SDA-cross-linked human serum albumin (HSA) served as a test sample, yielding over all methods and in triplicate analysis in total 2602 matched PSMs and 1390 linked residue pairs at 5% false discovery rate, as confirmed by the crystal structure. HCD wins in number of matched peptide-spectrum-matches (958 PSMs) and identified links (446). CID is most complementary, increasing the number of identified links by 13% (58 links). HCD wins together with EThcD in cross-link site calling precision, with approximately 62% of sites having adjacent backbone cleavages that unambiguously locate the link in both peptides, without assuming any cross-linker preference for amino acids. Overall quality of spectra, as judged by sequence coverage of both peptides, is best for EThcD for the majority of peptides. Sequence coverage might be of particular importance for complex samples, for which we propose a data dependent decision tree, else HCD is the method of choice. The mass spectrometric raw data has been deposited in PRIDE (PXD003737).